BIO-SAXS
Some examples are reported to illustrate the potential of small angle X-ray scattering (SAXS) in the study of biological systems
Characterization of a purified sample of biological macromolecules:
average size and shape, oligomeric state, quality assessment
SAXS is used to determine sizes and shapes of macromolecules and macromolecular assemblies in solution (such as protein-protein and protein-nucleic acid complexes, surfactant-protein complexes,...).
The initial slope and intercept of a SAXS profile are related (via the Guinier approximation) to the radius of gyration and the molecular mass of the particles in solution, respectively.
These simple and model-independent parameters can already provide useful information about the macromolecular average size and the oligomeric state of the macromolecule.
Example 1a: SAXS profiles (left) and Guinier fits (right) of the Human Serum Albumin monomer and dimer separated by size exclusion chromatography, as well as of the non-separated sample (mixture).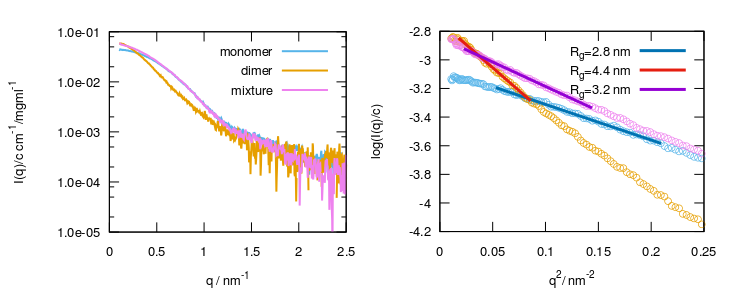
Characterization of flexible and disordered systems:
Kratky plot, ensemble analysis
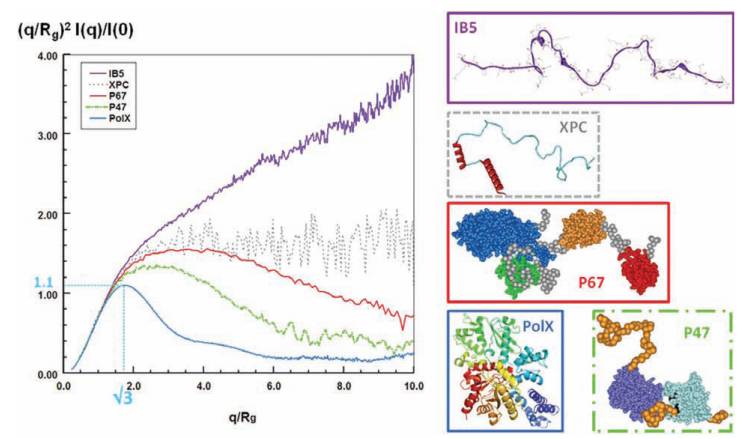
Intrinsically disordered proteins and multi-domain proteins with highly flexible linkers are difficult to characterize with high-resolution structural techniques. SAXS is a powerful tool to assess the flexibility of a macromolecular construct. The Kratky plot visualization of the scattering data (Intensity * scattering-vector^2 vs. scattering-vector) has a typical bell shape in the case of folded globular macromolecules, whereas it shows a plateau or a linearly increasing trend for flexible chains. The normalized version of the Kratky plot allows for the comparison between constructs of different size.
The scattering pattern of globular proteins in a normalized Kratky plot exhibits a bell-shaped profile with a clear maximum value of 1.104 for qRg=3, regardless of the size of the protein, and are all nearly superimposable in the q range 0<qRg<3. Conversely, for a random chain, the curve rises with increasing angle, to nearly reach a plateau between 1.5 and 2 and may further increase at q>0.2-0.3 Å-1, depending on the persistence length and the internal structure of the protein. In the figure: bell-shaped profile of a globular protein (PolX, blue line); curve of a protein consisting of several domains tethered by linkers with rather compact conformations (p47phox, dotted green line) or extended conformations (p67phox, continue red line); curve of a fully disordered protein with very short elements of secondary structure (XPC dotted grey line); and curve of a fully disordered and extended protein with short segments of polyproline repeats (salivary protein IB5, continue purple line).
A SAXS experiment complemented by modelling tools can reveal some information about the conformational ensembles populated by flexible constructs, assessing if fully extended or more compact arrangements are preferentially adopted, or if the coexistence of distinguishable conformations is likely.
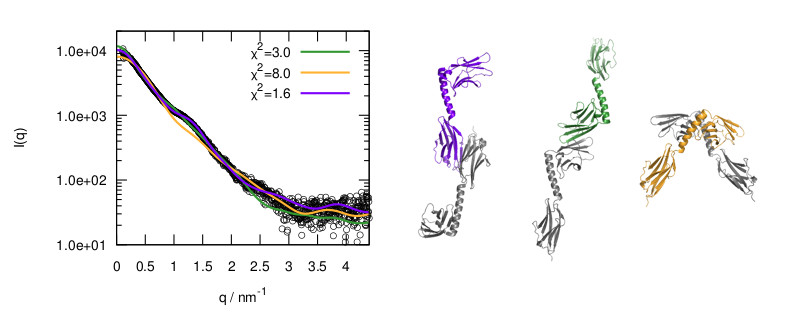
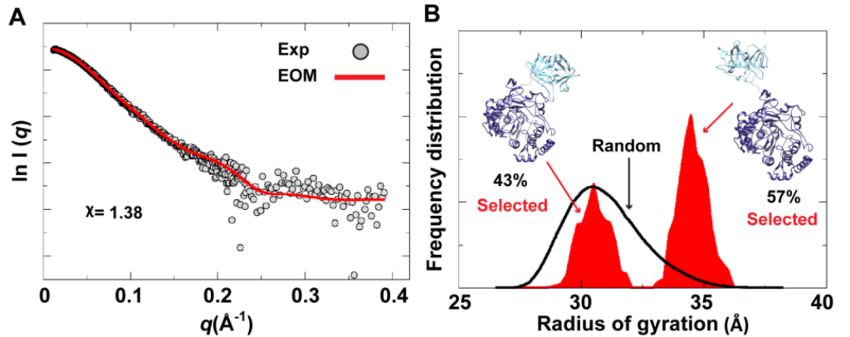
The Ensemble Optimization Method (EOM) analysis suggested the presence of two conformations for the poliovirus 3CD protein in solution. (A) The scattering profile calculated from the selected EOM sub-ensemble (red) is in good agreement with the experimental data (grey). (B) Shown is the EOM radius of gyration distribution (Rg) of the initial random pool (black line) and the selected sub-ensemble (red shades) for 3CD. The selected sub-ensemble corresponds to two conformers: an extended conformer that has no interdomain interactions and a compact conformer with interdomain interactions; the extended one is slightly preferred, contributing 57% to the total scattering. The SAXS experiments suggest that the conformation of 3CD protein observed in the crystal structure (extended) and that revealed by MD simulations (compact) co-exist in solution in almost equal proportions.
Study of processes depending on the environment:
oligomerization equilibria and conformational changes
One of the big advantages of SAXS compared to other techniques is the possibility to modify the solution environment with relative ease, in order to study the consequences of such changes at a macromolecular scale.
Indeed it is easy to probe protein conformational changes or oligomerization phenomena induced by pH change, addition of denaturants or ligand binding.
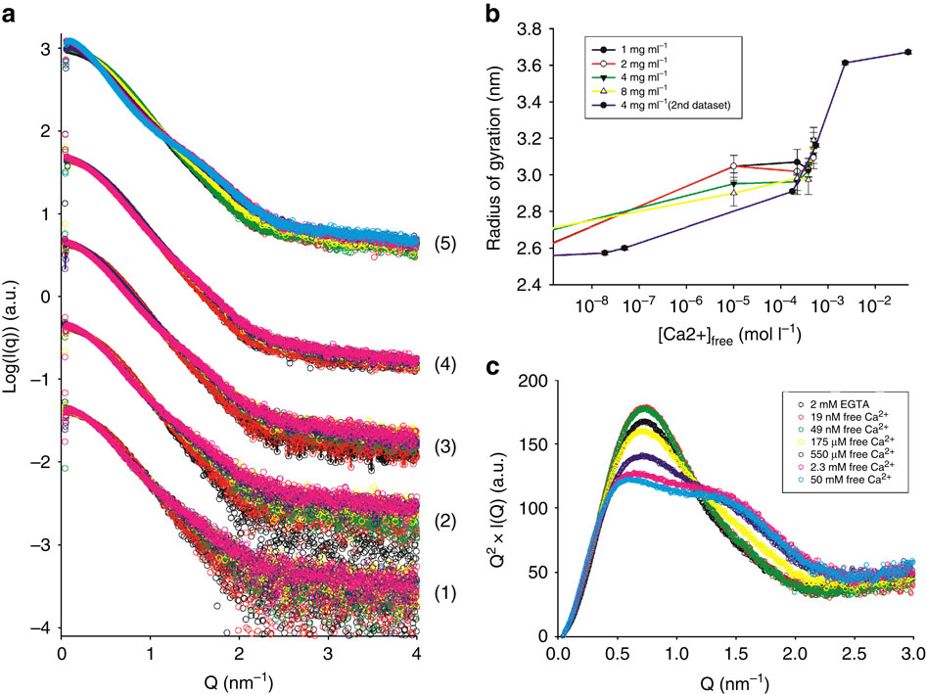
The calcium-regulated actin-binding protein adseverin undergoes a dynamic Ca2+-dependent equilibrium between inactive, intermediate and active conformations. Increasing calcium concentrations progressively shift this equilibrium from a main population of inactive conformation to the active form, which is less compact and more flexible as deduced from the increased radius of gyration and modified Kratky plot at high Ca2+ molarity.
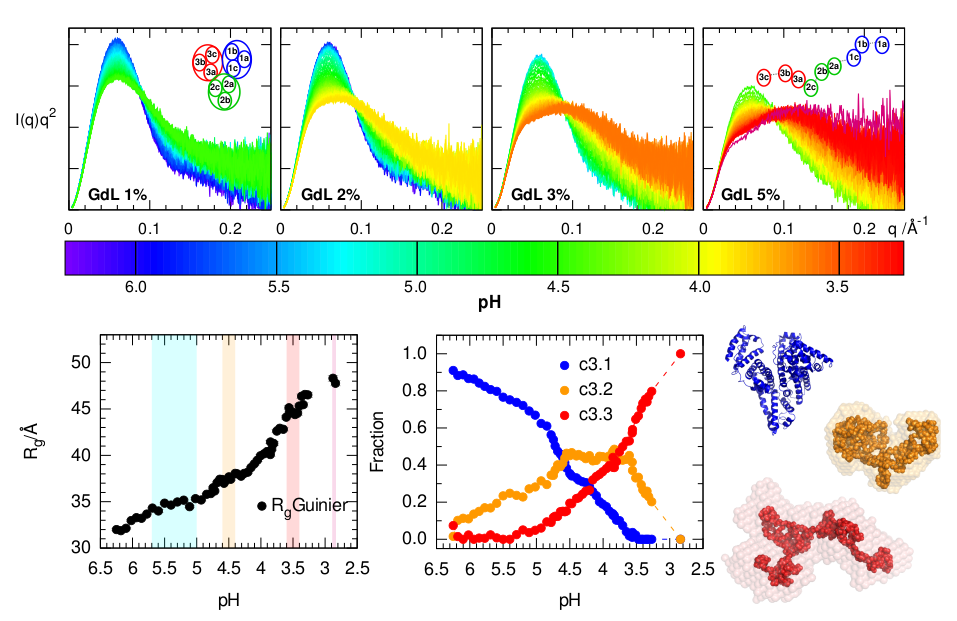
It is well known that Human Serum Albumin, the most abundant protein of body fluids, undergoes conformational changes in acidic conditions. The SAXS scattering profiles, especially when visualized in the form of a Kratky plot, are very sensitive to this acid-induced unfolding. A high sampling over the pH range achieved with a continuous acidification method helped discerning the conformers involved.
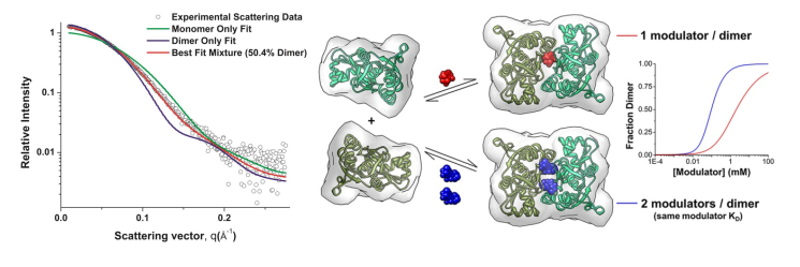
Example 3c: From C.P. Ptak, C.L. Hsieh, G.A. Weiland, R.E. Oswald, Role of stoichiometry in the dimer-stabilizing effect of AMPA receptor allosteric modulators, ACS Chem. Biol. 9 (2014) 128–133. doi:10.1021/cb4007166. (Further permissions related to the material excerpted should be directed to the ACS.)
Protein dimerization provides a mechanism for the modulation of cellular signaling events. In α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors, the rapidly desensitizing, activated state has been correlated with a weakly dimeric, glutamate-binding domain conformation. Allosteric modulators can form bridging interactions that stabilize the dimer interface. While most modulators can only bind to one position with a one modulator per dimer ratio, some thiazide-based modulators can bind to the interface in two symmetrical positions with a two modulator per dimer ratio. Based on SAXS experiments, dimerization curves for the isolated glutamate-binding domain of the AMPA receptor show that a second modulator binding site produces both an increase in positive cooperativity and a decrease in the EC50 for dimerization. Four body binding equilibrium models that incorporate a second dimer-stabilizing ligand were developed to fit the experimental data.
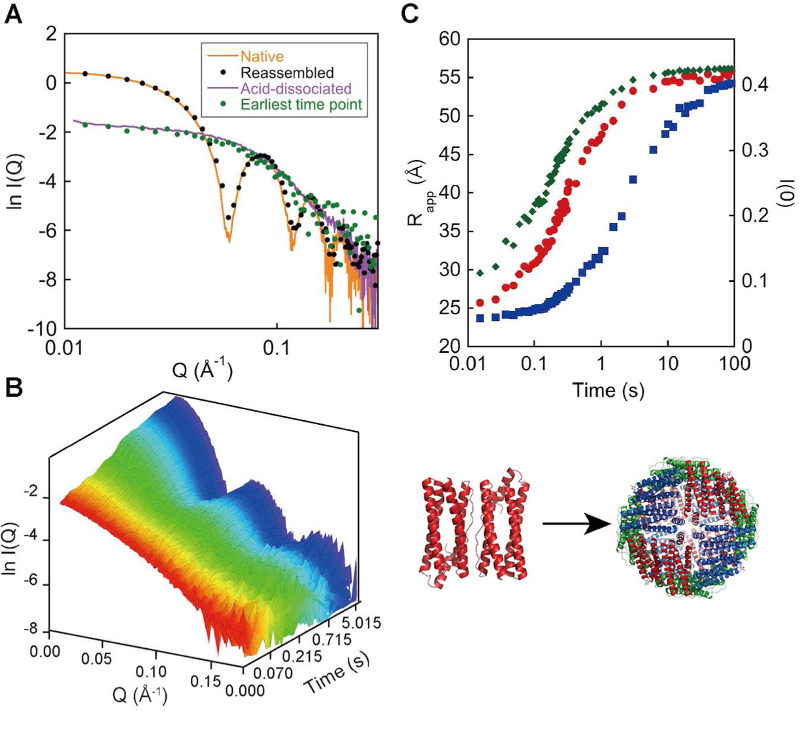
The assembly reaction of Escherichia coli ferritin A (EcFtnA) was studied using time-resolved SAXS. EcFtnA forms a cage-like structure that consists of 24 identical subunits and dissociates into dimers at acidic pH. The dimer maintains nativelike secondary and tertiary structures and is able to reassemble into a 24-mer when the pH is increased. The reassembly reaction was induced by a pH jump and followed by SAXS (A). Time-dependent changes in the forward scattering intensity (B, blue squares) and in the gyration radius (B, red circles) suggested the existence of a significant population of intermediate oligomers during the assembly reaction. The initial reaction was a mixture of second- and third-order reactions (formation of tetramers and hexamers) from the protein concentration dependence of the initial velocity. The time-dependent change in the SAXS profile was roughly explained by a simple model in which only tetramers, hexamers, and dodecamers were considered as intermediates.